Wespipeline¶
An implementation of a whole exome analysis pipeline using Luigi for workflow management.
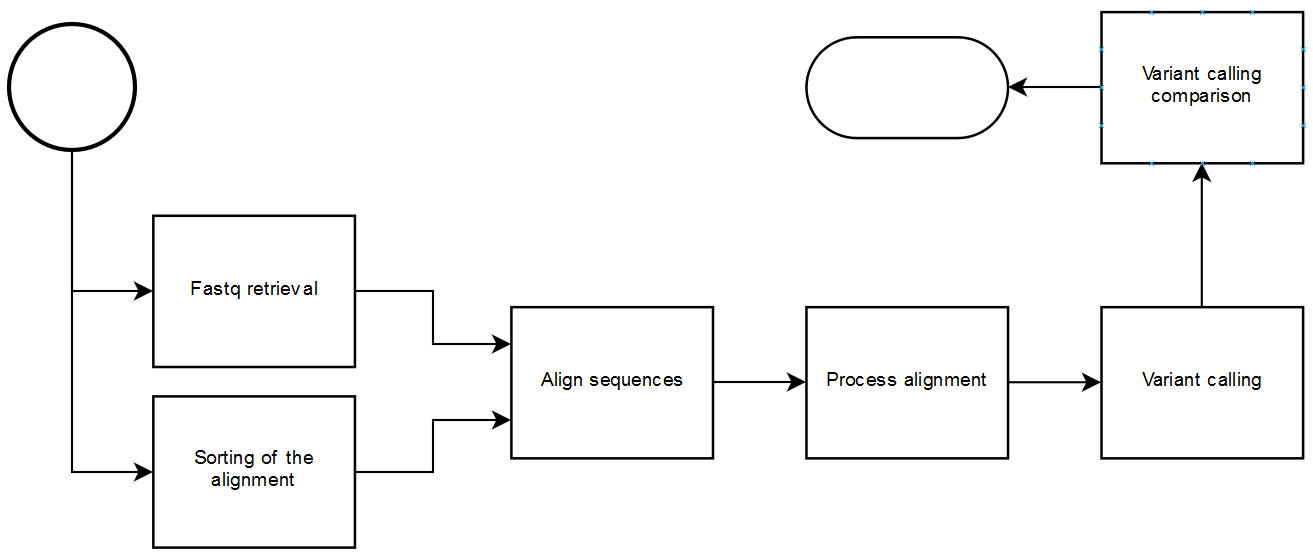
This package provides with the implementation of tasks for executing partial or complete variant calling analysis with the advantages of having a workflow manager: dependency resolution, execution planner, modularity, monitoring and historic.
Documentation for the latest version is being hosted by readthedocs
Installation¶
Wespipeline is available through pip, conda and manual installation. Install it from the package repositories
pip3 install wespipeline conda install -c jancho wespipeline, or download the project and build from source:
git clone https://github.com/Janchorizo/wespipeline.git && cd wespipeline && python3 setup.py install.
Notice that executing the analysis will involve different additional dependencies depending on the steps that executed and the parameters set for these. All possible are cited below and can be downloaded with the Anaconda distribution:
Secuence retrieval : Sra Toolkit, Fastqc
Reference genome retrieval : No needed dependency
Secuence alignment : Bwa
Alignment processing : Bwa Samtools,
Variant calling : Freebayes, Varscan, Gatk, Deepvariant
Variant calling evaluation : Vcf tools
In addition to the dependencies, conda can be used for installing the wespipeline package. An example for installing the miniconda distribution, the package and the dependencies is:
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda.sh
bash ~/miniconda.sh -b -p $HOME/miniconda
export PATH="$HOME/miniconda/bin:$PATH"
source $HOME/miniconda/bin/activate && \
conda config --add channels bioconda && \
conda config --add channels conda-forge && \
conda config --add channels jancho && \
conda install -y samtools && \
conda install -y bwa && \
conda install -y picard && \
conda install -y platypus-variant && \
conda install -y varscan && \
conda install -y freebayes && \
conda install -y fastqc && \
conda install -y sra-tools && \
conda install -y wespipeline
rm ~/miniconda.sh
Getting started¶
Installing or downloading the package will provide with a higher level task per step of the analysis, each of which can be executed in a similar fashion to other Luigi tasks.
Each of the six steps have a higher level task that can be scheduled in a similar fashion to other Luigi tasks:
python3 -m luigi --module wespipeline.<module> <Taskname> --<Taskname>-param value
Download the sequences using the NCBI accession number.
python3 -m luigi --module wespipeline.fastq FastqRetrieval \
--FastqRetrieval-paired-end true \
--FastqRetrieval-accession-number SRR9209557 \
--FastqRetrieval-create-report true
Or an external url.
python3 -m luigi --module wespipeline.fastq FastqRetrieval \
--FastqRetrieval-paired-end true \
--FastqRetrieval-compressed false \
--FastqRetrieval-accession-number SRR9209557 \
--FastqRetrieval-create-report true
Download the reference genome and create a report using FastqC.
python3.6 -m luigi --module tasks.reference ReferenceRetrieval
--workers 3 \
--ReferenceGenome-ref-url ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit \
--ReferenceGenome-from2bit True \
--GlobalParams-base-dir ./tfm_experiment \
--GlobalParams-log-dir .logs \
--GlobalParams-exp-name hg19
Or run the whole analysis, specifying the parameters for each of the steps.
python3 -m luigi --module tasks.vcf VariantCalling
--workers 3
--VariantCalling-use-platypus true
--VariantCalling-use-freebayes true
--VariantCalling-use-samtools false
--VariantCalling-use-gatk false
--VariantCalling-use-deepcalling false
--AlignProcessing-cpus 6
--FastqAlign-cpus 6
--FastqAlign-create-report True
--GetFastq-gz-compressed True
--GetFastq-fastq1-url ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/Garvan_NA12878_HG001_HiSeq_Exome/NIST7035_TAAGGCGA_L001_R1_001.fastq.gz
--GetFastq-fastq2-url ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/Garvan_NA12878_HG001_HiSeq_Exome/NIST7035_TAAGGCGA_L001_R2_001.fastq.gz
--GetFastq-from-ebi False
--GetFastq-paired-end True
--ReferenceGenomeRetrieval-ref-url ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit --ReferenceGenomeRetrieval-from2bit True
--GlobalParams-base-dir ./tfm_experiment
--GlobalParams-log-dir .logs
--GlobalParams-exp-name hg19
Tasks implemented¶
Module |
Task |
|---|---|
reference |
ReferenceGenomeRetrieval |
fastq |
FastqRetrieval |
align |
FastqAlignment |
processalign |
FastqProcessing |
variantcalling |
VariantCalling |
processalign |
VariantProcessing |
Acknowledgements¶
Special thanks to professor Luis Antonio Miguel Quintales for all the guidance and help provided during the development of this project.
Wespipeline : A whole exome secuencing variant calling pipeline¶
Using the pipeline
The analysis pipeline
Implementation